Reações orgânicas são
reações químicas envolvendo
compostos orgânicos. Os tipos básicos de reações da
química orgânica são
reações de adição,
reações de eliminação,
reações de substituição,
reações pericíclicas,
reações de rearranjo ou transposição e
reações redox. Em
síntese orgânica, reações orgânicas são usadas na construção de novas moléculas orgânicas. A produção de muitas substâncias pelo homem, tal como drogas, plásticos, a fabricação depende de reações orgânicas. As mais antigas reações orgânicas são a
combustão de combustíveis orgânicos e a
saponificação de gorduras para fazer
sabão. A moderna química orgânica inicia com a
síntese de Wöhler em
1828. Na história do
Prêmio Nobel de Química tem havido premiados pela invenção de reações orgânicas específicas tais como a
reação de Grignard em
1912, a
reação de Diels-Alder em
1950, a
reação de Wittig em
1979 e a
metátese de olefina em
2005.
Classificações
A química orgânica tem uma forte tradição de nomear uma reação específica em função do seu inventor ou inventores colaboradores e uma longa
lista de assim chamadas
reações nomeadas existe, conservadoramente estimadas em 1000. Um antiga reação nomeada é o
rearranjo de Claisen (1912) e uma recente é a
reação de Bingel (1993). Quando a reação nomeada é difícil de se pronunciar ou muito longa como a
reação de Corey-House-Posner-Whitesides ajuda usar uma abreviaçao como na
reação CBS. O número de reações apresentadas no atual processo é muito menor, por exemplo a
reação ene ou
reação aldólica. Outra abordagem para reações orgânicas é por tipo de
reagente orgânico, muitos deles
inorgânicos, requeridos em uma transformação específica. Os principais tipos são
agentes oxidantes tal como
tetróxido de ósmio,
agentes redutores tais como
hidreto de lítio e alumínio,
bases tais como
diisopropilamida de lítio e
ácidos tais como o
ácido sulfúrico.
Fundamentos
Os fatores que envolvem as reações orgânicas são essencialmente os mesmos de qualquer
reação química. Fatores específicos para as reações químicas são aqueles que determinam a estabilidade de reagentes e produtos tais como
conjugação,
hiperconjugação e
aromacidade e a presença e estabilidade de
reativos intermediários tais como
radicais livres,
carbocátions e
carbânions. Um composto orgânico pode cosistir de muitos
isômeros. Seletividade em termos de
regioseletividade,
diastereoseletividade e
enantioseletividade é consequentemente um importante critério para muitas reções orgânicas. A
estereoquímica de
reações pericíclicas é governada pelas
regras de Woodward-Hoffmann e as muitas
reações de eliminação pela
regra de Zaitsev. Reações orgãnicas são importntes na produção de
fármacos. Em uma revisão de 2006 estimou-se que 20% das conversões envolvendo
alquilações sobre átomos de nitrogênio e oxigênio, outras 20% envolvendo colocação e remoção de
grupos protetivos, 11% envolvendo formação de novas
ligações carbono-carbono e 10% envolvendo interconversões sobre
grupos funcionais.
Reações orgânicas por mecanismos
Não há limite para o número de reações orgãnicas possíveis e mecanismos. Entretanto, certos padrões são observado que podem ser usados para descrever muitas reaçõs comuns e úteis. Cada reação tem uma passo principal
mecanismo de reação que explica como ela acontece, através desta detalha descriçao de passos não é sempre clara de uma lista de reagentes isolados. Reações orgânicas podem ser organizadas em diversos tipos básicos. Algumas reações são classificáveis em mais de uma categoria. Por exemplo, algumas reações de substituição seguem uma marcha de adição-eliminação. Esta relação geral não pretende incluir cada reação orgânica. Entretanto, pretende-se cobrir as reações básicas.
- Adição eletrofílica ou EA
- Adição nucleofílica ou NA
- Adição radical ou RA
- Substituição nucleofílica alifática com mecanismos de reação SN1, SN2 e SNi
- Substituição nucleofílica aromática ou NAS
- Substituição nucleofílica acílica
- Substituição eletrofílica ou ES
- Substituição eletrofílica aromática ou EAS
- Substituição radical ou RS
Adição eletrofílica

Intermediário π de 2-elétrons-3-centros em uma adição eletrofílica a ligação dupla carbono-carbono

Intermediário σ de 2-elétrons-2-centros na adição eletrofílica de um íon H
+ a dupla ligação carbono-carbono
Em
mecanismos de reação em
química orgânica, uma reação de
adição eletrofílica (A
Ex ou Ad
Ex) é uma
reação de adição onde em um
composto químico, o substrato da reação, se perde um
ligação pi para permitir a formação de duas novas
ligações sigma. Nas reações de adição eletrofílica, os substratos mais comuns tem
ligações duplas ou
ligações triplas carbono-carbono.
- Y-Z + C=C → Y-C-C-Z
Adição nucleofílica
Adição nucleofílica é uma reação química, que ocorre em compostos carbonilados, onde ocorre a molécula com elétrons sobrando é ligada a uma parte pobre em elétrons (positiva) da molécula.
Reação de substituição
Numa reação de substituição, um grupo funcional numa molécula é substituído por outro. Em química orgânica, as reações de substituição são de grande importância. O conhecimento pormenorizado dos vários tipos de substituição possíveis permite prever o resultado de uma reação química, assim como escolher a temperatura e o solvente correctos para a sua ocorrência. Essas reações podem ocorrer de diversas maneiras, mas os tipos mais comuns são halogenação, sulfonação, nitração, alquilação e a acilação.
Substituição nucleofílica
Em química, uma sustituição nucleofílica ou nucleófila é um tipo de reação de substituição na que um nucleófilo, "rico em elétrons", substitui em uma posição eletrófila, "pobre em elétrons", de uma molécula a um átomo ou grupo, denominados grupo lábil. É um tipo de reação fundamental em química orgânica, onde a reação se produz sobre um carbono eletrófilo. Ainda que reações de substituição nucleofílica também podem ter lugar sobre compostos inorgânicos covalentes. Se ignoramos as cargas formais, em química orgânica a reação geral de substituição nucleofílica consiste em: - Nu: + R-L → R-Nu + L:
O nucléofilo Nu, mediante seu par de elétrons (
:), substitui no
substrato R-L, onde R é o eletrófilo, ao grupo saliente L, o qual leva consigo um par de elétrons. O nucléofilo pode ser uma
espécie neutra ou um
ânion, ainda que o substrato pode ser neutro ou têm carga positiva.
Um exemplo de substituição nucleófila é a
hidrólise de um
brometo de
alquilo, R-Br, sob condições
alcalinas, onde o nucleófilo é o
OH− e o grupo saliente é o Br
−.
- R-Br + OH− → R-OH + Br−
As reações de substituição nucleofílica são frequentes em química orgânica, e podem ser categorizadas de forma geral segundo tenham lugar sobre um carbono
saturado ou sobre um carbono
aromático ou
insaturado.
Substituições nucleófilas sobre carbonos saturados
Reações SN2 e SN1
Ao estudar-se as reações de substituição nucleófila em
haletos de alquila e compostos relacionados se observou que tinham lugar dois tipos de
mecanismo de reação. Os dois mecanismos são o
SN1 e o
SN2, onde S significa substituição, N simboliza nucleófilo e o número representa a
ordem de reação. A reação S
N2 (substituição nucleófila bimolecular) tem lugar em uma única etapa na que a adição do nucléofilo e a eliminação do grupo saliente se produz simultaneamente. Portanto é uma
reação concertada. A S
N2 é favorecida quando a posição do átomo de carbono eletrófilo é facilmente acessível ao nucleófilo. Por outro lado a reação S
N1 (substituição nucleófila unimolecular) implica duas etapas. Na primeira tem lugar a saída do grupo saliente e a formação do
intermediário carbocátion (etapa determinante da velocidade) e, na continuação, na segunda, o nucleófilo se une a este. A S
N1 tende a ser importante quando o átomo de carbono do substrato está rodeado de grupos volumosos, devido tanto a que tais grupos interferem
estericamente com a reação S
N2 como a que os carbonos mais substituídos formam carbocátions mais estáveis.
Reações de substituições nucleófilas
Existem muitas reações em química orgânica que implicam neste tipo de mecanismos. Exemplos habituais incluem:
-
- R-X → R-H usando LiAlH4 (SN2)
- Reações de hidrólises tais como:
-
- R-Br + OH− → R-OH + Br− (SN2) ou
-
- R-Br + H2O → R-OH + HBr (SN1)
-
- R-Br + −OR' → R-OR' + Br− (SN2)
Substituições nucleófilas sobre carbonos insaturados
A substituição nucleófila via mecanismos S
N1 ou S
N2 não tem lugar com haletos de
arilo ou
vinilo, ou compostos relacionados. Sob certas condições podem chegar a produzir-se substituições nucleófilas através de outros mecanismos (ver
substituição nucleofílica aromática). Quando a substituição ocorre no
grupo carbonilo, o
grupo acilo sofre o que se conhece como uma
substituição nucleofílica acílica. Este é o modo normal de substituição com derivados de
ácido carboxílico tais como
haletos de acilo,
anidridos carboxílicos,
ésteres ou
amidas.
Substituição nucleofílica aromática
Uma
substituição nucleofílica aromática é um tipo de reação de
substituição nucleofílica na qual o
nucleófilo desloca a um
grupo lábil, como um
haleto, em um
anel aromático.
Substituição nucleofílica acílica
A
substituição nucleofílica acílica consiste em uma reação de
substituição nucleofílica sobre a posição
carbonílica de um composto
acílico por parte de um
nucleófilo. Os compostos acílicos são derivados de
ácidos carboxílicos, o que incluem
ésteres,
amidas,
anidridos carboxílicos e
haletos de acilo. Os nucleófilos, nesta reação, incluem, por exemplo, reativos
aniônicos como
alcóxidos e
enolatos ou
bases como as
aminas.
Substituição eletrofílica
Reações de substituição eletrofílica são reações químicas nas quais um
eletrófilo substitui outro grupo, tipicamente mas não sempre
hidrogênio. A substituição eletrofílica é característica dos
compostos aromáticos. A
substituição eletrofílica aromática é um importante meio de introdução de grupos funcionais em anéis de
benzeno. A outra reação similar principal é a
substituição eletrofílica alifática.
Substituição eletrofílica aromática
As mais importantes das reações deste tipo são as
nitrações,
halogenações,
sulfonações aromáticas e as acilações e alquilações pelas
reações de Friedel-Crafts.
Substituição eletrofílica alifática
Em substituição eletrofílica em compostos
alifáticos, um
eletrófilo substitui um
grupo funcional. Esta reação é similar a
substituição nucleofílica alifática onde o reactante é um
nucleófilo mais que um
eletrófilo. Os dois mecanismos de reação eletrofílica,
SE1 e
SE2 (
Substituição
Eletrofílica), são também similares à contrapartes nucleófilas [[S
N1|SN1]] e [[S
N2|SN2]]. No curso S
E1 de ação o substrato primeiro se ioniza em um
carbânion e um resíduo orgânico positivamente carregado. O carbânion então facilmente recombina-se com o
eletrófilo. O mecanismo de reação S
E2 tem um único
estado de transição no qual a antiga ligação e a recentemente formada estão ambas presentes.
Nitração
A
nitração é a introdução irreversível de um ou mais grupo nitro (NO
2) em uma molécula orgânica. O grupo nitro pode atacar um carbono para formar um nitrocomposto (alifático ou aromático), um oxigênio para formar éster nitrado ou um nitrogênio para obter N-nitro compostos.
O sistema ácidosulfúrico/ácido nítrico, denominado
mistura sulfonítrica, é o reagente mais comum em nitração. Por exemplo, a nitração da glicerina seria da seguinte maneira: Aquecer o
ácido sulfúrico (
catalizador da reação ) juntamente com o composto que se quer nitrar, por exemplo,
glicerina (para formar
nitroglicerina). Após acrescenta-se lentamente, por gotejamento, o ácido nítrico.
A
nitração aromática consiste na substituição de um
hidrogênio do
anel aromático pelo NO
2, oriundo do
ácido nítrico.
As etapas de nitração do benzeno são:
(1) 2H
2SO
4 + HNO
3 → 2HSO
41- + NO
2+ + H
3O
+
(2) C
6H
6 + NO
2+ → C
6H
5NO
2 + H
+
(3) H
+ + H
3O
+ + 2HSO
41- → H
2O + 2H
2SO
4
Características da nitração
- Temperatura: Processos de nitração na grande maioria são reações exotérmicas e a temperatura influencia diretamente o curso da reação. Ao elevar a temperatura aumenta-se o grau de nitração, obtendo maior quantidade de produto nitrado, principalmente compostos supernitrados.
- Agitação: A reação de nitração pode ocorrer tanto na fase aquosa como na fase orgânica. Com agitação é possível que cada fase esteja sempre saturada pela outra, e, nestas condições, a velocidade de reação em cada fase é constante.
- Solubilidade: A medida em que se aumenta a temperatura, cresce a solubilidade dos nitrocompostos no ácido sulfúrico, e esta diminui com a diluição do ácido.
Sulfonação aromática
Sulfonação aromática é uma
reação orgânica na qual um átomo de
hidrogênio em um
areno é substituído por um
grupo funcional ácido sulfônico em uma
substituição eletrofílica aromática.
Os arenos (anéis aromáticos), sofrem reação de
sulfonação pela reação com o
ácido sulfúrico em alguns casos a frio e noutros a quente, com
ácido sulfúrico fumegante, uma mistura de
H2SO4 e
SO3. O
eletrófilo reativo pode ser tanto o o SO
3 quanto o HSO
3+ formado pelo equilíbrio:

Este poderá absorver água e formar HSO
3+ eletrofílico. Tal reagente sulfonador específico depende das condições de reação. A reação de sulfonação também ocorre por um mecanismo de várias etapas similares as reações de
nitração e
bromação. É de se observar, entretanto, que a reação de sulfonação é reversível, dependendo das condições da reação. A sulfonação é favorecida na presença de ácidos fortes, mas a dessulfonação é favorecida em solução ácida diluída e a quente.
Aplicações
Ácidos sulfônicos aromáticos são intermediários na preparação de
corantes e muitos
fármacos.
A sulfonação de
anilina produz
ácido p-aminobenzenosulfônico ou
ácido sulfanílico o qual é um
zwitterion com um incomum
ponto de fusão alto. A amida deste composto e compostos relacionados formam um grande grupo de medicamentos chamados
sulfas. As sulfonações pode ser feitas desde anéis benzênicos simples, como do
tolueno produzindo o
ácido toluenossulfônico, até
polímeros incluindo múltiplos anéis
benzênicos, como a sulfonação de
poliestireno pode ser usada para preparar o
poliestireno sulfonato de sódio.
Reação de Friedel-Crafts
As
reações de Friedel-Crafts (que podem ser
alquilação de Friedel-Crafts ou
acilação de Friedel-Crafts, conforme o tipo) são um conjunto de reações de
substituicão eletrófila aromática descobertas no ano de
1877 pelo
químico francês Charles Friedel e pelo químico
norte-americano James M. Crafts.
Tanto a
acilação como a
alquilação de Friedel-Crafts envolvem o uso do
cloreto de alumínio (
AlCl3) (e mais raramente do
cloreto de ferro III (
FeCl3) como ativador do
eletrófilo. No caso da alquilação, o eletrófilo é um
carbocátion (R+). No primeiro passo, o carbocátion é formado pela reação de um
cloreto de alquila com o
ácido de Lewis. Outros
haletos de alquila, como
brometos,
fluoretos ou
iodetos, também podem ser utilizados.
Haletos de vinila ou de
arila não funcionam, pois seus carbocátions são muito instáveis.
O produto da reação - um
alquilbenzeno - é mais reativo do que o próprio
benzeno. Para evitar a alquilação de um benzeno já alquilado, então, utiliza-se um excesso de benzeno na reação. Neste caso, o carbocátion tem mais chance de encontrar uma molécula de benzeno não-substituído, e formam-se, preferencialmente, apenas benzenos mono-substituídos.
Alquilação de Friedel-Crafts
Exemplo de alquilação de Friedel-Crafts.
Acilação de Friedel-Crafts
Exemplo de acilação de Friedel-Crafts. Ainda que não apareça, requer tratamento aquoso final.
Oxidação
Oxidação de álcoois
A oxidação de álcoois fornece
ácidos orgânicos quando se trata de álcoois primários,
cetonas quando são oxidados álcoois secundários e álcoois terciários não se oxidam. Pode-se também oxidar um álcool primário a
aldeído usando-se
cloro-cromato de piridina (PCC). Em laboratórios os agentes oxidantes mais utilizados são o
KMnO4 e o
K2Cr2O7.
Oxidação de álcoois primários
Em contato com
agentes oxidantes, os
álcoois primários reagem (oxidam) formando primeiro um
aldeído e então, com o
aldeído sendo oxidado, um
ácido carboxílico. Quando o
produto da oxidação de um álcool primário é um
aldeído ela é chamada normalmente de
oxidação branda; por sua vez, quando o produto é um
ácido carboxílico é chamada normalmente de
oxidação enérgica, porém esses nomes podem variar. A oxidação de um
álcool a
aldeído ou
cetona é também chamada de
desidrogenação (ou seja, perda de
hidrogênio).
Observe o exemplo abaixo:
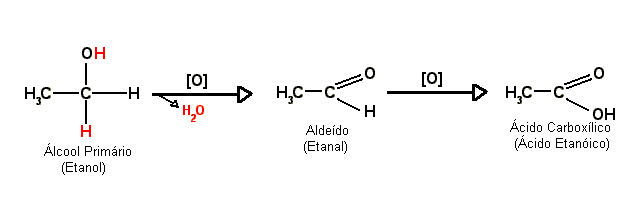
- O símbolo [O] significa oxidação.
- A flecha apotando para baixo com o H2O na ponta indica a água que foi produto da reação, ou seja, a reação também gerou água que foi retirada.
No esquema os
hidrogênios em vermelho reagiram com o oxigênio do
agente oxidante formando a
água que foi retirada. Portanto, diz-se que a oxidação é um processo de "retirada de hidrogênio".
Chamamos o processo inverso da oxidação de
redução e é representado por [H]. Sendo assim, a redução de um
ácido carboxílico gerará um
aldeído e a redução deste gerará um
álcoois primários. (Veja o esquema geral logo abaixo.)
Resumindo temos que a oxidação branda de um
álcoois primários gerará um
aldeído e a oxidação energica do mesmo gerará um
ácido carboxílico.

Oxidação de álcoois secundários
Os
álcoois secundários, diferente dos
primários, são oxidados de apenas uma forma gerando sempre como produto uma
cetona.
Veja este exemplo:

Oxidação de álcoois terciários
Não ocorre a oxidação de
álcoois terciários (exceto em uma reação de combustão total).
Oxidação de Alcenos
Os
alcenos também sofrem reações de oxidação que acontecem de três formas: oxidação branda, oxidação energética e ozonólise.
Oxidação branda
A oxidação branda (também conhecida como hidroxilação do
alceno), é uma reação feita com
agente oxidante que causa uma quebra na
dupla ligação do
alceno e a entrada de duas
hidroxilas (OH) formando assim um
diálcool. Um
agente oxidante muito usado neste caso é o
permanganato de potássio (KMnO
4).
Veja o exemplo abaixo:

Oxidação enérgica
Na oxidação enérgica, o agente oxidante "quebra" a
molécula na
dupla ligação e caso se forme um
aldeído ele é oxidado a
ácido carboxílico. Os agentes oxidantes mais comuns nesse caso são o
dicromato de potássio (K
2Cr
2O
7) e o
permanganato de potássio (KMnO
4).
Veja o exemplo abaixo:

Sempre que um composto que tem a
dupla ligação na ponta da
cadeia sofre oxidação enérgica um dos produtos que se formam é o
ácido carbônico (H
2CO
3) que por ser um
ácido instável transforma-se em
gás carbônico (CO
2) e
água (H
2O).
Ozonólise
A ozonólise é a quebra de um
alceno causada pelo
ozônio O
3 gerando como produtos uma
cetona e/ou um
aldeído. Esta reação deve ser realizada na presença de
água e de pó de
zinco. A molécula do
alceno é quebrada na
dupla ligação e dois átomos de
oxigênio (O) se adicionam.
Veja o exemplo abaixo:

O
peróxido de hidrogênio (H
2O
2) é destruido pelo pó de zinco para que não reaja com o
aldeído, formando hidróxido de zinco.




















